Was die Kraftwerke der Zelle in Form hält
Ein Team aus Deutschland und der Schweiz um Professor Oliver Daumke vom MDC hat untersucht, wie ein Protein der Dynamin-Familie die innere Membran der Mitochondrien verformt.
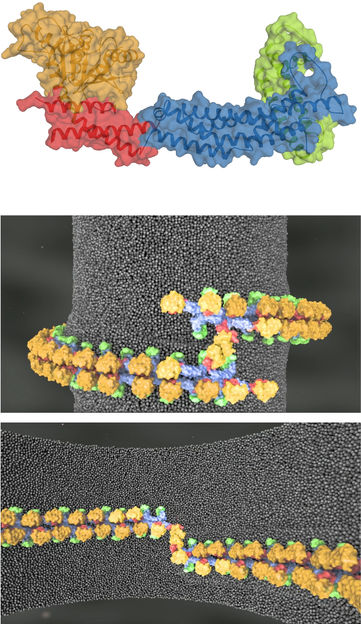
Mitochondrien sind alles andere als starre Gebilde. Sie teilen sich ständig und fusionieren wieder miteinander. Gesteuert wird das unter anderem von einem Protein aus der Dynamin-Familie namens Mgm1. Mithilfe der Kristallographie konnte das Team den dreidimensionalen Aufbau des Proteins beschreiben: den Motor (die GTPase-Domäne, orange), den Hebel (die BSE-Domäne, rot), den Stiel (blau) und das neu entdeckte Paddel (grün). Damit konnten molekulare Modelle entwickelt werden, wie Mgm 1-Stränge Membranen stabilisieren und umbauen können.
Daumke Lab, MDC
Mitochondrien sind die Kraftwerke unserer Zellen. Hier wird Energie in Form chemischer Verbindungen wie ATP gewonnen. Um dieser Aufgabe optimal nachgehen zu können, haben Mitochondrien einen ganz bestimmten Aufbau: Sie besitzen neben einer äußeren Membran auch eine innere, die mit zahlreichen Einstülpungen versehen ist. An dieser inneren Membran befinden sich alle Enzyme, die für die ATP-Produktion wichtig sind.
Mitochondrien sind keine starren Gebilde
„Uns hat interessiert, wie die Mitochondrien ihre charakteristische Form erhalten“, sagt Professor Oliver Daumke, der Leiter der MDC-Arbeitsgruppe „Strukturbiologie Membran-assoziierter Prozesse“ und einer der beiden Letztautoren der in „Nature“ veröffentlichten Studie. „Mitochondrien sind nämlich alles andere als starre Gebilde: Sie sind permanent damit beschäftigt, sich zu teilen und wieder miteinander zu fusionieren.“ Dabei werden zum Beispiel schadhafte Abschnitte zuverlässig entfernt.
„Aber nicht immer funktionieren die Mitochondrien so perfekt“, erläutert Dr. Katja Fälber von der Kristallographie-Abteilung des MDC, die eine von zwei Erstautorinnen der Studie ist. „Viele neurodegenerative Erkrankungen wie Alzheimer oder Parkinson entstehen unter anderem deshalb, weil das Gleichgewicht zwischen Teilung und Fusion der Mitochondrien gestört ist und die Nervenzellen deshalb langsam absterben“, sagt die Forscherin.
Eine molekulare Maschine zum Umbau der Membran
Gesteuert werden die stetigen Umwandlungsprozesse der Mitochondrien unter anderem von einem Protein aus der Dynamin-Familie. In der Hefe ist es Mgm1 (Mitochondrial genome maintenance 1 protein), beim Menschen OPA1 (Optic atrophy protein 1). Mutationen in dem Gen für OPA1 führen zu einer angeborenen Erkrankung des Sehnervs, der optischen Atrophie. Sie ist eine der häufigsten Ursachen der vererbten Blindheit.
„Sowohl Mgm1 als auch OPA1 sitzen auf der inneren Mitochondrien-Membran - und zwar insbesondere im Bereich der charakteristischen Einstülpungen “, erläutert Oliver Daumke. Dort fungieren die Proteine als molekulare Maschinen: Sie verwandeln chemische in mechanische Energie.
Bereits vor der aktuellen Studie war bekannt, dass die Proteine aus mehreren Untereinheiten bestehen: aus der GTPase-Domäne, die den eigentlichen Motor darstellt, aus der BSE-Domäne (bundle signalling element), die als Hebel agiert, und aus einem Stiel. „Uns hat insbesondere der Stiel interessiert, da er die Anordnung von Mgm1 in strangförmige Strukturen ermöglicht“, sagt Oliver Daumke. Es sind diese Stränge, die die Membran verformen. Ohne sie funktioniert die Maschine nicht.
Auf mehreren Wegen zu einem molekularen Modell
Um genauer herauszufinden, wie Mgm1 die Einstülpungen der inneren Membran stabilisiert und den permanenten Umbau der Mitochondrien-Membranen steuert, untersuchten die Forscher am MDC den dreidimensionalen Aufbau des Proteins per Kristallographie. „Mithilfe dieser Methode erhielten wir ein atomares Modell von Mgm1“, erklärt Katja Fälber.
Ergänzt wurde die Arbeit des Berliner Strukturbiologie-Teams von der Gruppe von Professor Werner Kühlbrandt am Max-Planck-Institut für Biophysik in Frankfurt am Main, die das Protein per Kryoelektronenmikroskopie unter die Lupe nahm. „Die Auflösung ist bei diesem Verfahren zwar geringer, doch dafür lässt sich das Protein anders als bei der Kristallographie auch im membrangebundenen Zustand untersuchen“, erläutert die MDC-Wissenschaftlerin.
Auf diese Weise stießen die Forscher gemeinsam auf eine vierte, bislang unbekannte Domäne von Mgm1, die sie das „Paddel“ nannten. „Dabei handelt es sich um eine langgestreckte Untereinheit, über die sich das Protein an die innere Membran der Mitochondrien heftet“, berichtet Oliver Daumke. „Darüber hinaus haben wir im Stiel des Proteins diejenigen Kontaktflächen ermittelt, die für die Anordnung von Mgm1 in Stränge erforderlich sind.“ Damit hatte das Team alle Puzzleteile zusammen, um die Prozesse an der Membran am Computer zu simulieren.
Mutationen im Mgm1 führen zum Zerfall der Mitochondrien
Um zu überprüfen, ob die identifizierten Flächen für die Stabilisierung der inneren Mitochondrien-Membran wirklich entscheidend sind, tauschte eine Gruppe um Professor Martin van der Laan vom Universitätsklinikum des Saarlandes in Homburg in diesen Regionen einzelne Aminosäuren aus. „Und tatsächlich verlor das Protein dadurch seine Funktion“, sagt Oliver Daumke: „Die Mitochondrien konnten ihre charakteristischen Einstülpungen nicht mehr richtig ausbilden und auch nicht mehr miteinander fusionieren.“ Zurückgeblieben seien viele einzelne Mitochondrien-Fragmente.
In einer weiteren Kollaboration mit einem Team um Professor Aurel Roux von der Universität Genf konnten die Forscher schließlich mittels Fluoreszenzmikroskopie live beobachten, wie sich Mgm1 an Membranen heftet. „Überraschend und neu war dabei die Beobachtung, dass sich das Protein nicht nur an die Außenseite von künstlichen Membranröhrchen anlagert, sondern auch an deren Innenseite“, sagt Katja Fälber. „Diese Geometrie entspricht der der Mitochondrien-Einstülpungen und wurde bisher für kein anderes Protein aus der Dynamin-Familie beschrieben.“
Ziel ist auch, die genetische Blindheit besser zu verstehen
Das Team hofft nun, dass sich die jetzt gewonnenen Erkenntnisse irgendwann auch medizinisch nutzen lassen werden. „Die Ergebnisse unserer Studie erklären, was genau bei der Entstehung einer optischen Atrophie im Auge schiefläuft – wie Mutationen im OPA1-Gen also bewirken, dass das Protein nicht mehr richtig funktioniert“, sagt Oliver Daumke. Vielleicht lasse sich die Krankheit mit diesem Wissen eines Tages sogar heilen.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News




Weitere News von unseren anderen Portalen






























Verwandte Inhalte finden Sie in den Themenwelten
Themenwelt Fluoreszenzmikroskopie
Die Fluoreszenzmikroskopie hat die Life Sciences, Biotechnologie und Pharmazie revolutioniert. Mit ihrer Fähigkeit, spezifische Moleküle und Strukturen in Zellen und Geweben durch fluoreszierende Marker sichtbar zu machen, bietet sie einzigartige Einblicke auf molekularer und zellulärer Ebene. Durch ihre hohe Sensitivität und Auflösung erleichtert die Fluoreszenzmikroskopie das Verständnis komplexer biologischer Prozesse und treibt Innovationen in Therapie und Diagnostik voran.
Themenwelt Fluoreszenzmikroskopie
Die Fluoreszenzmikroskopie hat die Life Sciences, Biotechnologie und Pharmazie revolutioniert. Mit ihrer Fähigkeit, spezifische Moleküle und Strukturen in Zellen und Geweben durch fluoreszierende Marker sichtbar zu machen, bietet sie einzigartige Einblicke auf molekularer und zellulärer Ebene. Durch ihre hohe Sensitivität und Auflösung erleichtert die Fluoreszenzmikroskopie das Verständnis komplexer biologischer Prozesse und treibt Innovationen in Therapie und Diagnostik voran.