Künstliche Intelligenz gegen den Fehlerteufel
Einsatz künstlicher Intelligenz zur Fehlerkorrektur bei Einzelzellenanalysen
Moderne technische Möglichkeiten erlauben es, einzelne Zellen zu sequenzieren und jeweils individuell herauszufinden, welche Gene gerade abgelesen werden. Diese Methoden sind sehr fein, dadurch aber auch sehr fehleranfällig: Geräte, Umwelt aber auch die Biologie selbst können für Ausfälle und Unterschiede zwischen den Messungen verantwortlich sein. Forscher des Helmholtz Zentrums München haben gemeinsam mit Kollegen der Technischen Universität München (TUM) und des englischen Wellcome Sanger Institute nun Algorithmen entwickelt, die diese Fehlerquellen berechen- und korrigierbar machen.
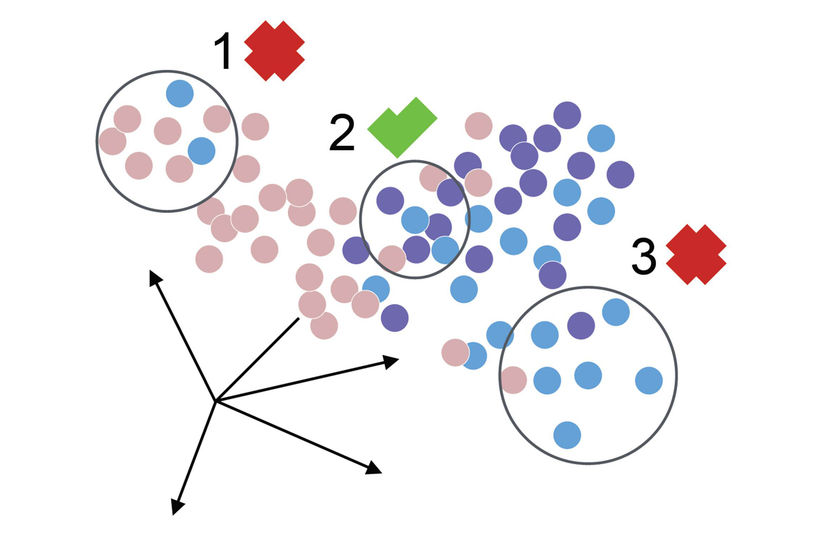
Modified from Büttner, M. et al. (2019)
Es ist ein visionäres Vorhaben von enormen Ausmaßen – das Human Cell Atlas-Projekt kartiert alle Gewebe des menschlichen Körpers zu verschiedenen Zeitpunkten mit dem Ziel eine Referenzdatenbank zur Entwicklung personalisierter Medizin zu schaffen, also ‚gesunde‘ und ‚kranke‘ Zellen vergleichen zu können. Möglich wird das durch sogenannte Einzelzell-RNA-Sequenzierung – also vereinfacht gesagt: die Möglichkeit, nachzuvollziehen, welche Gene diese winzigsten Bausteine des Lebens gerade an- oder ausschalten. „Das ist methodisch gesehen ein enormer Sprung, denn früher waren solche Daten immer nur aus großen Gruppen von Zellen zu gewinnen, weil die Messungen so viel RNA benötigten“, erklärt Maren Büttner. „Die Ergebnisse waren also immer nur der Mittelwert aller eingesetzter Zellen, heute bekommen wir für jede einzelne Zelle exakte Daten“, so die Doktorandin am Institute of Computational Biology (ICB) des Helmholtz Zentrums München.
Durch die feineren Messungen steigt allerdings auch die Anfälligkeit für den sogenannten Batch-Effekt. „Dabei handelt es sich um Abweichungen zwischen mehreren Messungen, die beispielsweise bereits entstehen können, wenn die Temperatur des Gerätes leicht abweicht oder sich die Verarbeitungszeit der Zellen verändert“, erklärt Maren Büttner. Zwar gäbe es hier verschiedene Modelle, um den Fehler herauszurechnen, allerdings sind diese Methoden stark davon abhängig, wie groß der Effekt eigentlich ist. „Um das herauszufinden, haben wir eine nutzerfreundliches, robustes und sensitives Maß namens kBET entwickelt, dass Unterschiede zwischen Experimenten quantifiziert und damit verschiedene Korrektur-Ergebnisse vergleichbar macht“, sagt Büttner.
Neben dem Batch-Effekt sind sogenannte Null-Messungen (englisch: dropout events) bei der Einzelzellsequenzierung eine große Herausforderung. „Wir sequenzieren also eine Zelle und stellen fest, dass ein bestimmtes Gen in dieser Zelle überhaupt kein Signal von sich gibt“, veranschaulicht ICB-Direktor Prof. Dr. Dr. Fabian Theis. „Dahinter kann sich nun ein biologischer oder ein technischer Grund verbergen: Entweder wird das Gen nicht abgelesen, weil es in diesem Moment schlicht keine Rolle spielt, oder aber die Sequenz ist aus technischen Gründen nicht erfasst worden“, so der Professor für Mathematische Modellierung biologischer Systeme an der TUM.
Um diese Fälle zu erkennen, nutzten die Bioinformatiker Gökcen Eraslan und Lukas Simon aus Theis‘ Gruppe die große Anzahl der Datenpunkte und entwickelten einen sogenannten Deep Learning Algorithmus. Dabei handelt es sich um künstliche Intelligenz, die Lernprozesse simuliert, wie sie auch beim Menschen vorkommen (neuronale Netze).
Über ein neues Wahrscheinlichkeitsmodell und Vergleich der ursprünglichen und rekonstruierten Daten ermittelt der Algorithmus, ob in diesem Fall ein biologischer oder ein technischer Ausfall zugrunde liegt. „Durch dieses Modell lassen sich sogar Zelltyp-spezifische Korrekturen ermitteln, ohne dass sich zwei unterschiedliche Zelltypen künstlich ähnlicher werden“, so Fabian Theis. „Als einer der ersten Deep Learning Methoden im Bereich Einzelzell-Genomik hat der Algorithmus den weiteren Vorteil, gut auf Datensätze mit Millionen von Zellen zu skalieren.“
Eines – das ist den Wissenschaftlern wichtig – ist die Methode aber nicht: „Wir bauen hier keine Software, um Ergebnisse beliebig zu ‚glätten‘. Unser Ziel ist es vor allem, Fehler ausfindig zu machen und zu korrigieren“, so Fabian Theis. „Mit diesen möglichst korrekten Daten können wir dann in den Austausch mit unseren Kollegen weltweit gehen und unsere Ergebnisse mit ihren vergleichen.“ Beispielsweise, wenn die Helmholtz-Forscher ihren Anteil für den Human Cell Atlas beisteuern, denn gerade hier ist die Verlässlichkeit und die Vergleichbarkeit der Daten von größter Wichtigkeit.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News







Weitere News von unseren anderen Portalen






























Verwandte Inhalte finden Sie in den Themenwelten
Themenwelt Zellanalyse
Die Zellanalyse ermöglicht es uns, Zellen in ihren vielfältigen Facetten zu erforschen und zu verstehen. Von der Einzelzellanalyse über die Durchflusszytometrie bis hin zur Bildgebungstechnologie – die Zellanalyse bietet uns wertvolle Einblicke in die Struktur, Funktion und Interaktion von Zellen. Ob in der Medizin, der biologischen Forschung oder der Pharmakologie – die Zellanalyse revolutioniert unser Verständnis von Krankheiten, Entwicklung und Behandlungsmöglichkeiten.
Themenwelt Zellanalyse
Die Zellanalyse ermöglicht es uns, Zellen in ihren vielfältigen Facetten zu erforschen und zu verstehen. Von der Einzelzellanalyse über die Durchflusszytometrie bis hin zur Bildgebungstechnologie – die Zellanalyse bietet uns wertvolle Einblicke in die Struktur, Funktion und Interaktion von Zellen. Ob in der Medizin, der biologischen Forschung oder der Pharmakologie – die Zellanalyse revolutioniert unser Verständnis von Krankheiten, Entwicklung und Behandlungsmöglichkeiten.